Polymer recycling unveiled: Supercomputing and molecular dynamics paving the way to circular economy

Published on 19/12/2023
A polymer is a substance or material consisting of very large molecules composed of many repeating subunits [1]. The large molecules are made by bonding (chemically linking).
We live in an era dominated by polymers. You can find polymers in your plastic water bottle or sandwich box, shopping bags, or the nylon and polyester in your sneakers. We often use and dispose of plastics, negatively affecting the environment.
Mats Denayer is a PhD student at the General Chemistry Research Group (ALGC) of Vrije Universiteit Brussel. In his PhD, he developed a new computer-based protocol to predict whether polymers will dissolve in certain liquids (solvents).
Proven valid, the program can be used for industrial purposes such as polymer recycling. Mats' research fits within the context of transitioning from our current linear "use and dispose" economy to a circular economy through the development of (polymer) recycling processes, as the European Union desires.
Understanding Polymer Dissolution through Molecular Dynamics Simulations
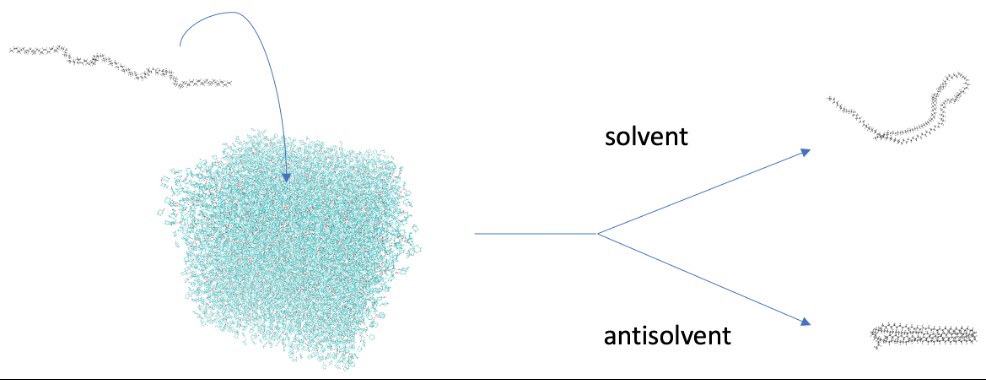
Instead of doing traditional experiments in the lab, Mats uses a technique called classical molecular dynamic simulations to study how polymers behave when put in a liquid. He zooms in on what happens at the atomic level, and the simulations help him understand the different steps in the dissolution process.
Mats Denayer (ALGC-VUB): “We look at polymer swelling [2], solvent penetration and chain disentanglement [3]. We also check how the shape of the polymer changes during this process by measuring how much of its surface is touching the liquid. All this information helps us figure out how well the polymer can dissolve in that particular liquid. It's like trying to understand a puzzle piece by piece to see if it fits into a bigger picture.”
For his simulations, Mats uses the Tier-1 supercomputing infrastructure of the Vlaams Supercomputer Centrum (VSC) for two key reasons.
Mats Denayer (ALGC-VUB): “Firstly, when it comes to simulating systems with a vast number of atoms, such as one to two million in our case, time becomes a critical factor. We explicitly model every polymer and solvent atom, making these simulations incredibly large. Running these simulations on standard computers or Tier 2 systems would take too long. Tier-1 supercomputers offer the computational power we need to tackle such complex simulations efficiently. Secondly, we aim to explore a large number of polymer/solvent combinations, ultimately translating into a high-throughput screening process.
Jan Turek (HPC Business developer and consultant Vlaams Supercomputing Centrum): “The more factors that are considered in simulations, the closer we get to a realistic depiction, minimising the need for approximations and inching towards the concept of a digital twin. Industry partners prefer realism, as it fosters trust. The goal is to provide as accurate a representation as possible to align with the industry's demands."
Scaling Polymer Methodology with LUMI
Using supercomputing, particularly its GPU partition, has allowed Mats to scale up and evaluate the methodology on many different materials.
Mats Denayer (ALGC-VUB): “Initially, we just started with simulating only one polymer chain in a known (anti-)solvent, and then we built a protocol around it. Then, we used the Tier-1 system to run our computational procedure on unknown polymer and solvent combinations involving multiple polymer chains. In the end, we modelled four different polymers in twelve solvents up till now.”
As the demand for computational power increased with the growing complexity of systems to be calculated, Mats recognised the necessity to scale up from Tier-1 to LUMI, Europe's fastest Tier-0 supercomputer. Mats Denayer (ALGC-VUB): “With LUMI, due to the high availability of GPU nodes, we could harness the power of 4 GPUs simultaneously for a single simulation, significantly accelerating our calculations.”
Window opened on other applications
And there is more: the protocol is not restricted to polymer recycling but to any application examining the affinity between certain molecules. Mats Denayer (ALGC-VUB): “We are now working on adapting our protocol to be used in other fields like polymer-assisted drug delivery. We are looking into the affinity of a polymer and a solvent. Polymer-assisted drug delivery looks into the affinity between a polymer and a drug. There is a lot of similarity.”
Benefits
The computational protocol developed by ALGC-VUB is a viable tool to predict the behaviour of a polymer in a liquid, and it is also a viable tool to understand observed polymer-solvent interactions.
The protocol is being developed so that it can be used very easily by the industry. It works like a black box: industrial users can just give the input (a polymer) and ask which solvent to dissolve or precipitate. Or the input could also be: ‘I have polymer A and solvent B, and the output will tell what will happen to the polymer. The industrial user doesn’t have to know and does not get to see the calculations needed to give the result.
Supercomputing elevating research in chemistry

Mats Denayer (ALGC-VUB): “I really recommend supercomputing to anyone active in the field of computational chemistry. It has helped us tremendously in speeding up our calculations and scaling up the size of our systems. Without it, performing a standard simulation of 50 ns on a system of 2 million atoms would not be feasible as the simulation time would be prohibitively large. For illustration, on a normal laptop, the simulation would take more than two years, whereas the supercomputer GPU units get the job done in 15 days.”
Jan Turek (Vlaams Supercomputing Centrum): “Employing molecular dynamic simulations allows for an in-depth exploration of the intricate details, enabling a comprehensive analysis of the ongoing processes. In this context, supercomputing offers a valuable opportunity to elevate our research. As we continue pushing the boundaries, there is still much more to discover."
[1] Definition source: https://en.wikipedia.org/wiki/Polymer
[2] How the polymer gets bigger or ‘swells’ as it starts to mix with the liquid
[3] How the liquid starts to go inside the polymer and how the polymer's long chain of atoms starts to untangle itself
Relevant links:
- https://youtu.be/6xqQc-P8vxc?t=207
- Presentation Mats Denayer at the VSC User Day 2022
- M. Denayer, J. Vekeman, F. Tielens, F. De Proft, Physical Chemistry Chemical Physics (PCCP), 23, 25374-25387, 2021, 3.676
- W. Stuyck, K. Janssens, M. Denayer, F. De Schouwer, R. Coeck, K.V. Bernaerts, J. Vekeman, F. De Proft, Green Chemistry, 24, 6923-6930, 2022, IF: 11.034


After completing his Chemistry education at the VUB in 2019, Mats Denayer joined the ALGC group as a predoctoral researcher supervised by Frank De Proft in October of the same year. His work as a PhD student is part of an SBO (Strategic Basic research) project at the Research Foundation Flanders, in which a computational and interpretive method for fast and accurate polymer solubility prediction by combining classical molecular dynamics simulations with non-covalent interaction (NCI) analysis is developed.